INTRODUCTION
Acetaminophen (APAP, Tylenol®, also known as paracetamol, N-acetyl-p-aminophenol) has analgesic and antipyretic properties, and it is considered to be safe in therapeutic doses. However, overdose of APAP can lead to severe liver injury both in humans and in experimental animals [1,2]. Investigations into the mechanisms underlying APAP toxicity have revealed a metabolic conversion of APAP into a highly reactive intermediate, N-acetyl-p-benzoquinonimine (NAPQI), by cytochrome p450s (CYPs), which are abundant enzymes in liver cells. The reactive NAPQI metabolite can be detoxified by glutathione, it binds to the functional protein, or it initiates the formation of reactive oxygen species (ROS) to trigger lipid peroxidation. This peroxidation is a critical factor that causes destruction and damage to cell membranes and finally leads to necrosis or apoptosis of the liver cells [3]. Although it is believed to be safe at therapeutic doses, the number of intoxications by APAP seems to be increasing. According to the American Association of Poison Control Centers, nearly 140,000 poisoning cases were reported in 2006 alone, in which more than 100 patients died [4].
Although liver toxicity by APAP has been widely investigated, studies on non-hepatic toxicity, such as cardiotoxicity, have not been conducted sufficiently. Thum and Borlak showed the presence of metabolic enzymes (CYPs) in the heart and suggested the possibility of metabolic activation of APAP [5]. However, there have been a few reports on cardiotoxicity that showed myocardial damage with sub-endocardial hemorrhage and focal necrosis following an acetaminophen overdose [6,7]. It is uncertain whether these changes are direct cardiotoxic effects or secondary changes due to shock or hepatic failure, and little is known about the molecular events occurring within the myocardium following APAP insult.
Therefore, the objectives of the present study were to identify the alterations in gene expression in cultured cardiomyocytes, H9C2 cells treated with APAP, to evaluate the possible toxic mechanism in the heart.
MATERIALS AND METHODS
I. Materials
H9C2 cells, which are rat cardiomyocytes, were purchased from the Korean cell line bank (Seoul, Korea). The growth properties of the H9C2 cells used in this study are as adherent cells. Acetaminophen, dulbecco's modified eagle's medium (DMEM), 3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), tris, ethylene diamine tetraacetic acid (EDTA), and agarose were purchased from Sigma-Aldrich (MO, USA). Trypsin, antibiotics, and fetal bovine serum (FBS) were purchased from GIBCO (Invitrogen Corp, NY, USA). The RNA isolation kit (RNAgents®) was purchased from Promega Company (WI, USA). The AccuPower RT/PCR premix for the amplification of nucleic acid was purchased from Bioneer (Daejeon, Korea). The DNA chip was purchased from Affymetrix (Affymetrix Gene Chip, Rat Gene 1.0 ST Array, CA, USA). APAP was dissolved in a 0.5% (w/v) carboxymethyl cellulose solution containing 0.1% Tween 80.
II. Cell Culture
H9C2 cells were maintained in DMEM containing 10% heat-inactivated FBS, penicillin 100 IU/mL, and streptomycin 10 µg/mL. Cells were grown and maintained in 28-cm2 cell culture flasks at 37℃ in a 5% CO2 humidified incubator. For the cytotoxicity test, cells were cultivated using 96-well plates.
III. Cell Viability Test
Cell viability was measured by the MTT assay. Cells were seeded on 96-well tissue culture plates with 5×103 to 2×104 cells in 100 µL media per well. After 24 hours stabilization of the cells, they were treated with 1, 2.5, 5, 10, and 20 mM concentrations of APAP for 24, 48, 72, and 96 hours, respectively. Control cells were treated with the vehicle only. At the end of the exposure, 40 µL of MTT solution (2 mg/mL) was added, and the cells were incubated for 4 hours at 37℃. The cells were treated with 150 µL of DMSO, and absorbance was quantified in 540 nm using the microplate spectrophotometer system (VersaMax, Molecular Devices, Sunnyvale, CA, USA). The viability of the treated group was expressed as a percentage of the control group, which was assumed to be 100%. The cell viability test was performed more than three times for each concentration and exposure time of APAP.
IV. APAP Treatment, RNA Isolation, and Gene Expression Profiling
The cells were incubated with 10 mM concentration of APAP for the designated times (6, 12, and 24 hours), and a negative control culture was prepared for the exposure duration of 6, 12, and 24 hours. Total RNA was isolated using an RNA isolation kit as instructed by the manufacturer (Promega Company, Madison, WI, USA). RNA quality was assessed with an Agilent 2100 bioanalyzer using the RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, The Netherlands), and quantity was determined by ND-1000 Spectrophotometer (NanoDrop Technologies Inc., DE, USA). The 260/280 nm ratio of total RNA used in the study was 1.8-2.0. For each RNA sample, 300 ng of total RNA was converted to double-stranded cDNA, as recommended by the manufacturer. Using a random hexamer incorporating a T7 promoter, amplified RNA (cRNA) was generated from the double-stranded cDNA template though an in vitro transcription reaction and purified with the Affymetrix sample cleanup module. The corresponding cDNA was regenerated through a random-primed reverse transcription, using a deoxyribonucleotide triphosphate mix containing deoxyuridine triphosphate. The cDNA was then fragmented by uracil DNA glycosylase and apurinic/apyrimidinic endonucleasse 1 restriction endonucleases and end-labeled by terminal transferase reaction incorporating a biotinylated dideoxynucleotide. Fragmented end-labeled cDNA was hybridized to the GeneChip® rat Gene 1.0 ST arrays for 16 hours at 45℃ and 60 rpm, as described in the Gene Chip Whole Transcript Sense Target Labeling Assay Manual (Affymetrix). After hybridization, the chips were stained and washed in a GeneChip® Fluidics Station 450 (Affymetrix) and scanned with a GeneChip® Array Scanner 3000 7G (Affymetrix). Affymetrix data were extracted, normalized, and summarized with the robust multi-average method implemented in the Affymetrix Expression Console. The fold change of a probe was calculated by dividing the signal value of the APAP-treated cells by that of the control cells. The signal log ratio (log 2 ratio) and the change call (increase, decrease, or no-change) were calculated. Gene expression profiles with a signal log ratio of 1 or more, or -1 or less (i.e., ≥ two-fold change) were taken to find significantly deregulated genes. The results were classified using hierarchical and K-mean clustering algorithms implemented in TMEV software 4.0. (The Institute for Genomic research, Rockville, MD, USA). Differentially expressed genes were then classified based on their known biological functions using the Database for Annotation, Visualization, and Integrated Discovery software. Functional classification was based on both gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis.
V. RT-PCR Analysis
RNAs were isolated from cultured cardiomyocytes treated with 1, 5, 10, and 20 mM acetaminophen for 24 hours, respectively. Reverse transcription-polymerase chain reaction (RT-PCR) analysis was performed to confirm the expression of heme oxygenase 1 (Hmox1), glutathione-S-transferase (Gst), thioredoxin reductase (Txnrd), and actin. RT-PCR was performed using oligo deoxythymidine primer in 20 µL volumes at 42℃ for 60 minutes. The RT-PCR reaction was performed with 1 µg of total RNA, 1 µL of 20 µM oligo dT primer, and 18 µL of reaction mixture, which was provided by AccuPower RT/PCR PreMix (Bioneer, Daejeon, Korea). Then, PCR was performed in a 20 µL total mixture volume for 25-28 cycles at 95℃ for 1 minute, 55℃ for 1 minute, 72℃ for 1 minute. Amplified cDNA products were separated on 1.5% agarose gel by electrophoresis. The primer sequences of amplified genes are shown in Table 1. Actin mRNA was also amplified as a loading control.
RESULTS
I. Cytotoxicity of APAP in Cultured H9C2 Cells
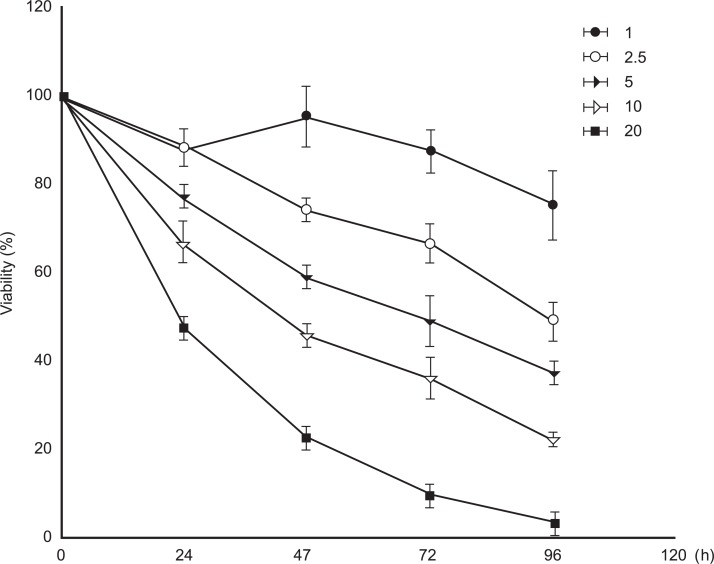
To examine the toxic effects, H9C2 cells were incubated with different concentrations (1, 2.5, 5, 10, and 20 mM) of APAP, and viability was determined at 24, 48, 72, and 96 hours after treatment, respectively. As shown in Figure 1, cell viability was decreased by treatment with APAP in a time- and dose-dependent manner. Results represent the means of three separate experiments, and error bars represent the standard error of the mean. Viability of the group treated with the 10 mM concentration of APAP was down to 60-70% of the control group at 24 hours after treatment, and further decreased to 40-50% of the control group at 48 hours after treatment. More than 50% of the cells treated with 20 mM APAP did not survive within 24 hours of exposure.
II. Gene Expression in APAP-Treated Cultured H9C2 Cells
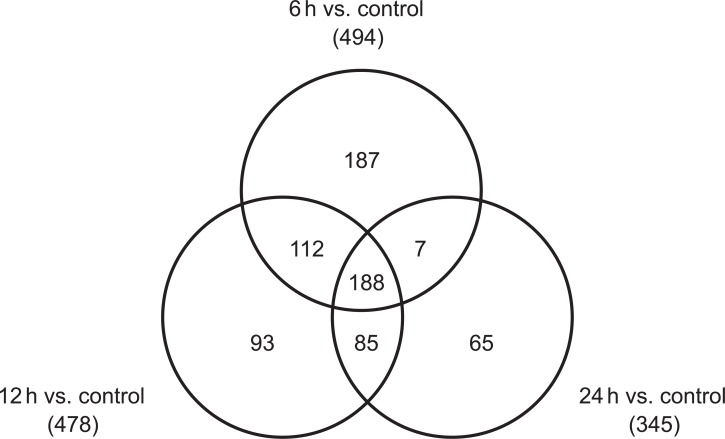
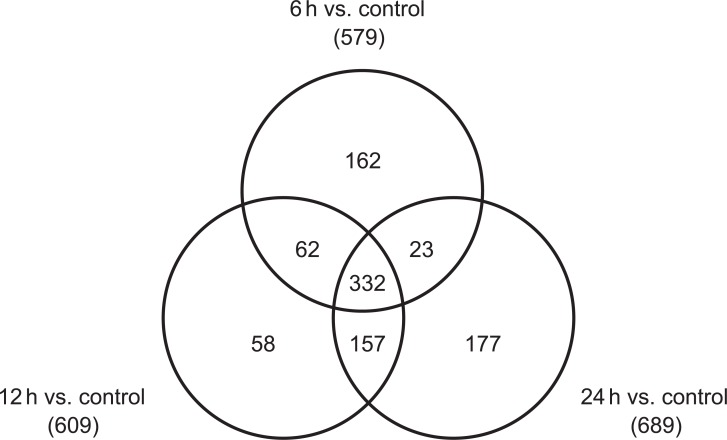
Using microarrays, more than 26,000 gene probes were analyzed in cultured H9C2 cells. Six hours after treatment with a 10 mM concentration of APAP, 494 genes were up-regulated, and 579 genes were down-regulated in cardiomyocytes. Meanwhile, 345 genes were up-regulated and 689 genes were down-regulated 24 hours after treatment. Figures 2 and 3 show the Venn diagrams for the up-regulated and down-regulated genes, respectively. Functional classification based on both gene ontology and KEGG pathway analysis showed that many biological functions were affected by APAP treatment in cultured cardiomyocytes, which include cellular process, purine and pyrimidine metabolism, regulation of cellular metabolic process, and regulation of gene expression.
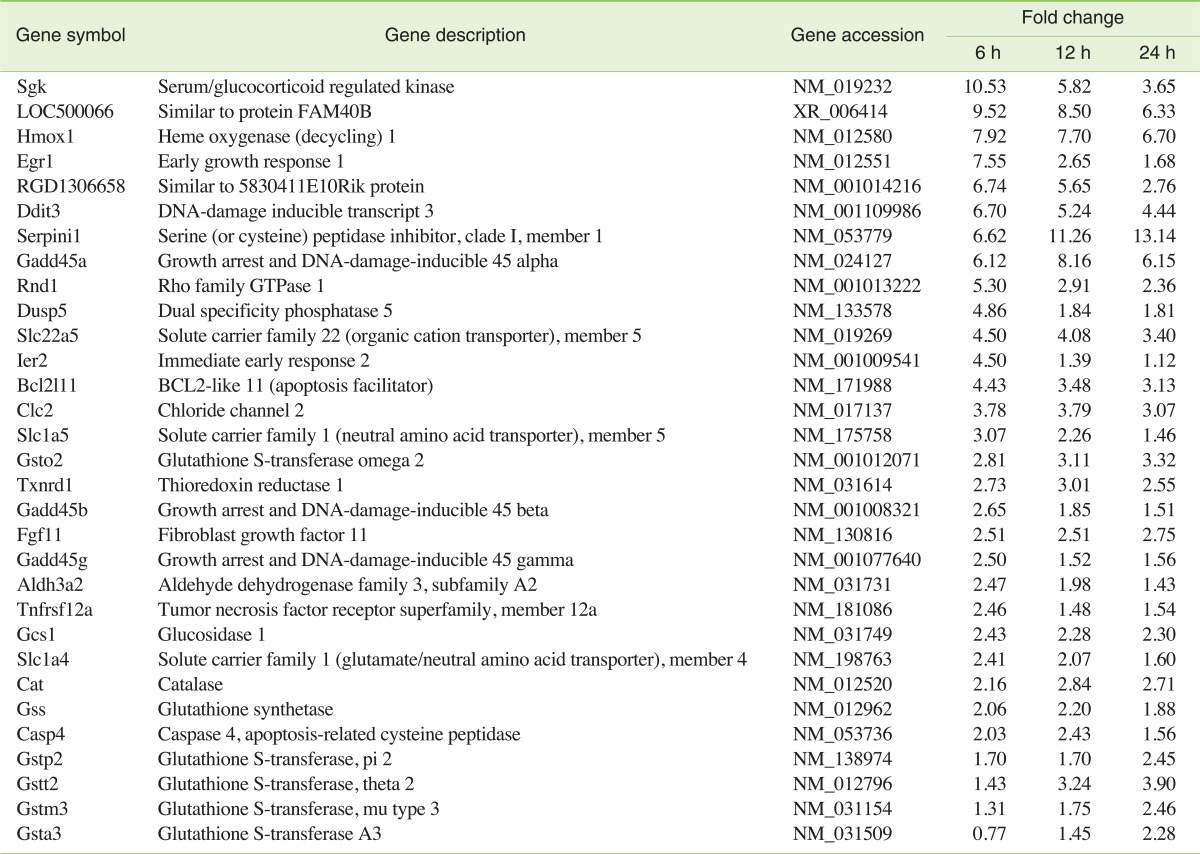
Tables 2 and 3 show the partial lists of genes induced and repressed by APAP, respectively. The fold changes of these genes are also included in these tables. The genes affected by APAP treatment were classified according to the related biological processes. In cultured cardiomyocytes treated with APAP, several oxidative stress-related genes were significantly up-regulated, and some of them may be an indication of protection against the deleterious effects of oxidative stress. Hmox1, an important enzymatic antioxidant system, was induced 7.9 fold at 6 hours, 7.7 fold at 12 hours, and 6.7 fold at 24 hours after treatment. The inductions of Thioredoxin reducctase 1 (Txnrd 1) and the catalase (Cat) gene were also observed during the time of exposure (Txrnrd 1: 2.7, 3.0, and 2.6 fold; cat: 2.2, 2.8, and 2.7 fold, respectively). Among the 17 probe sets annotated with Gst, the Glutathione S-transferase omega 2 (Gsto2) gene was induced during the entire period of exposure (2.8, 3.1, and 3.3 fold, respectively). Gstt2 was up-regulated at 12 and 24 hours after exposure, while Gsta3, Gstp2, and Gstm3 were up-regulated only at 24 hours after exposure. Glutathione synthetase (Gss) was up-regulated 6 and 12 hours after exposure. DNA damage and apoptosis-related genes were the other meaningful functional groups of genes. During the entire exposure period, the induction of DNA-damage-inducible transcript 3 (Ddit3) was observed (6.7, 5.2, and 4.4 fold, respectively). Concomitant down-regulations of ankyrin repeat and suppressor of cytokine signalling (SOCS) box-containing protein 2 (Asb2) were also observed (0.27, 0.22, and 0.35 fold during the entire exposure period, respectively).
APAP-treated cardiomyocytes also displayed a significant increase in expression of all three isoforms of growth arrest and DNA-damage-inducible 45 genes (Gadd45α, β, and γ). Among 11 investigated caspases, only caspase-4 was induced by 2.0 fold at 6 hours and by 2.4 fold at 12 hours after exposure. The induction of expression of Egr1, which is a nuclear transcription factor involved in the regulation of multiple genes, was rapid and transient, peaking at 6 hours (7.6 fold) and falling to near normal level by 24 hours after exposure to APAP. Chloride channel 2 (Clc2), a candidate for the native inwardly rectifying chloride channel, was induced during the entire period of exposure (3.8, 3.8, and 3.1 fold, respectively).
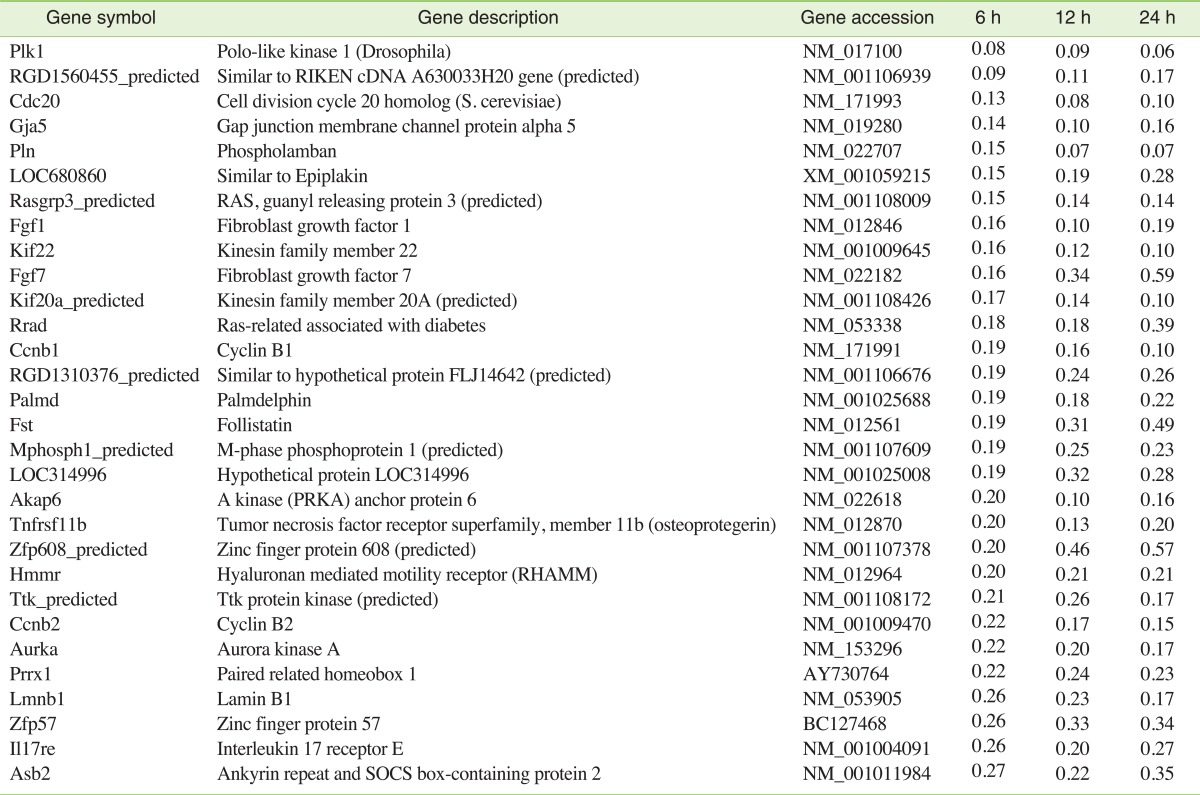
Meanwhile, cell proliferation-related genes were repressed by APAP. Cyclin B1 (Ccnb1), Cyclin B2 (Ccnb2), and cell division cycle 20 homolog (Cdc20) were repressed-cyclin B1: 0.19, 0.16, and 0.10 fold; cyclin B2: 0.22, 0.17, and 0.15 fold; and Cdc20: 0.13, 0.08, and 0.10 fold, by the exposure of 6, 12, and 24 hours, respectively. Among the 23 probe sets annotated with fibroblast growth factors (Fgfs), Fgf1 and Fgf7 were repressed (Fgf1: 0.16, 0.10, and 0.19 fold; Fgf7: 0.16, 0.34, and 0.59 fold, respectively). Gap junction membrane channel protein alpha 5 (Gja5) and phospholamban (Pln), which are involved in myocardial contraction, were repressed at all time points (Gja5: 0.14, 0.10, and 0.16 fold; Pln: 0.15, 0.07, and 0.07 fold, respectively). Palmdelphin (Palmd), a lipid raft-associated protein implicated in cell shape control, was also repressed at all time points after exposure to APAP (0.19, 0.18, and 0.22 fold, respectively).
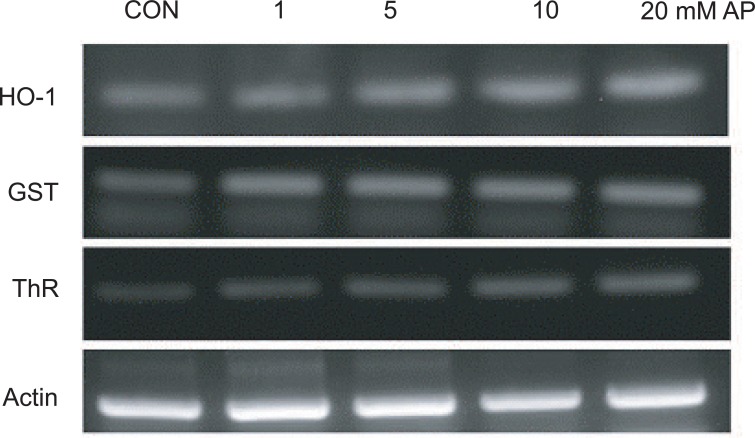
III. Verification of Gene Expression Data with RT-PCR
RT-PCR was performed after treatment with 1, 5, 10, and 20 mM APAP for 24 hours for the verification of microarray data. Three oxidative stress-related genes (Hmox1, Gst, and Txnrd) were chosen for verification. RT-PCR results of the expression patterns of the three genes after exposure to various concentrations of APAP are shown in Figure 4. RT-PCR analysis showed concentration-dependent increases in gene expression of the three genes, and these expression patterns were consistent with the gene expression results obtained from the microarray analysis.
DISCUSSION
Several previous reports have reported significant alterations in gene expression, encompassing a wide array of physiological processes in the livers of APAP-treated mice [8,9]. However, little is known about the adverse effects and pathogenic mechanisms occurring within the myocardium following APAP insult. Our findings provide the first evidence of APAP-induced alterations in gene expression in cardiomyocytes in vitro.
In this study, the concentration of APAP to see the gene expression was chosen based on the result of cytotoxicity test shown in Figure 1. Cultured cardiomyocytes were exposed to 10 mM APAP for 6, 12, and 24 hours, respectively. At the concentration, the viability of cells was slightly decreased to about 66% after 24 hours treatment. According to the viability graph, the mortality was significantly decreased by the decrease of exposure duration, which is proper to see the gene expression in sub-mortal level of APAP.
Dysregulation of normal gene expression may hold crucial clues about the pathogenic mechanisms of cytotoxicity and may be a sensitive indicator of potential adverse effects in the absence of the occurrence of overt toxicity. Previous investigations of the mechanisms underlying APAP toxicity have revealed a metabolic conversion of APAP to a highly reactive intermediate, NAPQI, by CYPs, which are abundant enzymes in liver cells [3]. Although CYP genes are mainly expressed in the liver, many CYP enzyme families have been identified in the heart [5]. H9C2 cell line is a commercially available myogenic cell line derived from embryonic rat heart ventricles. It was demonstrated that multiple CYPs were expressed in H9C2 cells at comparable levels to those expressed in the rat heart [10]. Therefore, this cell line offers a valuable in vitro model to study the direct toxic effects of APAP in the heart, excluding systemic effects derived from shock or hepatic failure. The present study revealed that numerous critical genes were affected in cardiomyocytes treated with APAP.
Oxidative stress-related genes(Hmox1, Txnrd1, Cat, Gst, and Gss)were significantly upregulated by APAP. Hmox1 is an important enzymatic antioxidant system [11]. In mammalian cells, Hmox1 is induced primarily at the transcriptional level by various types of oxidative stress [12,13]. Each by-product of its heme catabolism (carbon monoxide or bilirubin/biliverdin) mediates the antioxidant, anti-inflammatory, and anti-apoptotic effects of Hmox1 [14]. In addition, a combination of Hmox1 byproduct can act to protect the cell and tissue from further damage. The induction of Hmox1 gene expression by APAP in this study suggested an increase of the antioxidant system to the APAP-triggered oxidative stress. Thioredoxin (Trx) system, involving Trx and Txnrd, is a ubiquitous thiol oxidoreductase system that regulates cellular reduction/oxidation (redox) status [15]. Previous reports have suggested that Trx provides protection against ROS-mediated cardiotoxicity [15,16]. Catalase catalyzes the formation of oxygen and water from two hydrogen peroxide molecules. Over-expression of catalase and superoxide dismutase in cultured cells and whole animals has provided protection against the deleterious effects of oxidative stress [17]. The upregulation of Hmox1, Txnrd, and Cat may be a part of adaptive mechanisms to compensate for the dysregulated redox status and to protect against oxidative stress caused by APAP. Stimulation of glutathione synthesis and induction of glutathione S-transferases can protect cells from oxidative stress by conjugating or scavenging the oxidative electrophile molecules or ROS. The induction of Gst and Gss in cardiomyocytes is comparable to the responses in hepatocytes after exposure to toxic doses of APAP [18]. The induction of Gst and Gss also suggests a stimulated redox cycle in cardiomyocytes and may be a defensive measure to protect cells from oxidative stress provoked by APAP.
Some of the DNA damage and apoptosis-related genes were upregulated by APAP. Ddit3, also known as DNA damage-inducible gene 153 (Gadd153), plays a role in the regulation of cell cycle and apoptosis. Ddit3 is expressed ubiquitously and can be induced by a wide variety of cellular stresses, including DNA damage and oxidative stress [19]. Previous studies have reported that Gadd153 overexpression and the resulting down regulation of cardiac ankyrin repeat protein might have causative roles in the apoptosis of cardiac myocytes during hypoxia and ischemia-reperfusion injury [20]. In this study, Ddit3 over-expression and the concomitant down regulation of ankyrin repeat protein (Asb2) suggest the impact of oxidative stress and/or DNA damage, as well as the resulting progression of apoptotic processes in cultured cardiomyocytes after exposure to APAP. Although the exact function of Gadd45 is not yet clear, the proteins encoded by these genes have been reported to play pivotal roles as stress sensors that modulate and integrate the response of mammalian cells to a variety of environmental and physiological stressors: cell cycle arrest, DNA repair, cell survival, apoptosis, and senescence. The increased expression of Gadd45 genes may be a response to the cellular stress-inducing DNA damage provoked by APAP [21].
In cardiovascular cells, early growth response-1 (Egr1) is known to be a regulator that controls the expression of a wide variety of genes, including sodium-calcium exchanger-1 and sarco(endo)plasmic reticulum Ca2+-ATPase 2 (Serca2). Egr1 is involved in the cardiovascular pathological processes, such as atherosclerosis, ischemia-reperfusion, allograft rejection, and cardiac hypertrophy [22,23]. In this study, Egr1 mRNA expression in cardiomyocytes was rapid and transient, peaking at 6 hours after APAP exposure (7.6 fold) and falling to near normal levels by 24 hours after stimulation. It can be suggested that the rapidly and transiently induced Egr1 dynamically linked changes in the local cellular environment (high concentration of APAP in culture media) to the altered expression of genes mediating a broad spectrum of cellular pathologies. Clc2 belongs to a large CLC family of voltage-gated chloride channels. In mammalian cardiac cells, Clc2 has been proposed to be the molecular candidate for the native inwardly rectifying Clchannels. Clc2 is activated by hyperpolarization, extracellular acidification, and oxidative stress. In the heart, an increase in Clc2 conductance could be proarrhythmic under pathological conditions such as ischemia and hypoxia [24]. The association between Clc2 gene induction by APAP and the possibility of clinical pro-arrhythmic effect needs further investigation.
While the expression of many genes was observed in cardiomyocytes treated with APAP, others were down-regulated. Cell proliferation-related genes were repressed by APAP. Cyclin B1/CDK1 is necessary for mitosis to occur. Progression of a cell through the cell cycle is promoted by a number of cyclin-dependent kinases (CDKs), which drive the cell forward through the cell cycle. The ability of the mammalian cardiomyocyte to divide decreases significantly during development, which is consistent with a down-regulation in the activities and expressions of cyclin and CDK complexes. Overexpression of the cyclin B1/CDK1 complex re-initiates cell division in adult cardiomyocytes [25]. In this study, cyclin B1 was repressed by 0.1 fold at 24 hours after treatment, and the repressed expression of cyclin B1 might inhibit cardiomyocyte cell division. Cell division cycle 20 homolog (Cdc20), a cell-cycle related protein, is essential for the completion of mitosis. Cdc20 binds to and activates the ubiquitin ligase activity of the anaphase-promoting complex, promoting cell-cycle-regulated ubiquitination and proteolysis of a number of critical cellcycle-regulatory targets. Cdc20 also serves as an integrator of multiple intracellular signaling cascades that regulate progression through mitosis [26]. The transcriptional down-regulation of Cdc20 may have negative implications for the proper ordering of cell cycle events.
Expression of Fgfs and Fgf receptors by adult cardiac myocytes contributes to DNA synthesis and cell survival. In addition to their mitogenic function, Fgfs can participate as endogenous cardio-protective agents by increasing NO and cGMP levels in cardiomyocytes. cGMP inhibits the L-type calcium entry into the cardiomyocyte, preventing calcium accumulation in the mitochondrial matrix and cytosol within cardiomyocytes. Inhibition of intracellular calcium may shorten phase 2 of the cardiac action potential, providing a negative inotropic effect and decreasing myocardial oxygen and energy consumption. NO can inhibit apoptosis by nitrosylating active sites of cysteine residues in caspases [27]. The repressed expression of Fgf1 and Fgf7 may be the result of direct damage to the cultured cardiomyocytes by APAP.
Gap junction membrane channel protein alpha 5 (Gja5) plays important roles in myocardial contraction, and the gene expression was significantly decreased by APAP. A gap junction contains clusters of tens to thousands of intercellular channels called "gap junction channels," which allow passage of ions and small molecules; these junctions mediate intercellular signals. Gap junctions in the heart play an important functional role by electrically coupling cells, thereby organizing the pattern of current flow to allow coordinated muscle contraction. Cardiac gap junctions are therefore intimately involved in normal conduction, as well as in the genesis of potentially lethal arrhythmias [28] .
PLN is a transmembrane sarcoplasmic reticulum (SR) phosphoprotein that inhibits the activity of Ca2+ ATPase SERCA2a. The Ca2+ transport into the SR by SERCA2a accounts for at least 70% of the removal of Ca2+ from the cytosol during cardiac relaxation. PLN binds to SERCA2a and inhibits its activity by lowering its apparent affinity for Ca2+. As a result, PLN determines myocardial relaxation rates and influences cardiac contractility. PLN protein levels are correlated with cardiac contractile parameters and Pln mutations have been reported to impair SR Ca2+ cycling, leading to cardiac dysfunction and heart failure [29]. The repressed expression of Pln suggests the possibility of myocellular calcium dysregulation and impairment of relaxation and contractile function. Palmdelphin is a cytosolic isoform of paralemmin-1, a lipid raft-associated protein implicated in plasma membrane dynamics and cell shape control. Palmdelphin is expressed in variable abundances in nearly all tissues, with highest levels in the heart and lung. It associates with cytoskeleton-linked protein structures [30]. Down-regulation of palmdelphin may present difficulties in the maintenance of intracellular cytoskeleton-linked structures and cause deformation of the cultured cardiomyocytes. In this study, the gene expression of palmdelphin was significantly decreased by APAP.
In conclusion, this in vitro study revealed that numerous critical genes were affected by APAP in rat cardiomyocytes of H9C2 cells. Genes induced by APAP included those associated with oxidative stress, DNA damage, and apoptosis. Genes repressed by APAP included those associated with cell proliferation, myocardial contraction, and cell shape control. These results suggest that APAP may cause unfavorable molecular changes in the heart in vivo, and they present some vital clues to the molecular mechanisms of the cardiotoxicity of APAP.