Introduction
Di(2-ethylhexyl)phthalate (DEHP), which is commonly used to improve the flexibility of polyvinylchloride, such as building materials and medical products, is one of the most highly produced chemicals and is ubiquitous in the environment [1]. The exposure range of the general population, excluding medical and occupational exposure, has been estimated to be 5–100 µ g/kg body weight [2]. DEHP that enters the human body is rapidly converted to monoethylhexyl phthalate in the liver and gut [3-5] and then additionally hydrolyzed into secondary oxidized metabolites [4, 6]. Although DEHP has a relatively short half-life and is rapidly excreted through the urine or feces [6, 7], DEHP and its metabolites have been detected in the urine [8-10], serum [11], and breast milk [12] on repeated exposure.
Recent epidemiological studies have suggested that the levels of urinary phthalate metabolites in adults are positively associated with metabolic disorders, including abdominal obesity, insulin resistance [13], and non-alcoholic fatty liver disease (NAFLD) defined by the hepatic steatosis index [14-17]. Laboratory studies have provided supporting evidence showing that the disruption of the expression of lipogenesis and lipolysis enzymes after DEHP exposure in male rats [18-20] and liver cell lines [21-23] is involved in the pathogenic progression of NAFLD; however, the molecular mechanisms have not yet been clearly identified.
Two acyl-CoA:diacylglycerol acyltransferase (DGAT1 and DGAT2) enzymes, belonging to distinct gene families in mammals, are found in the liver [24]. Both enzymes are expressed in the liver and are involved in the final step of the conversion of free fatty acids to triglyceride (TG) [24, 25]. In patients with NAFLD, DGAT1 expression in the liver was increased two-fold compared with that in normal individuals [26]. In addition, Dgat1 deficient mice showed a reduction in TG levels and did not develop fatty liver after high-fat diet administration [27, 28]. It seems that DGAT enzymes have an important role in TG accumulation in the liver. Our previous study showed that perinatal exposure to DEHP induced hepatic lipid accumulation through mediated by an increase in Dgat1 expression in adult male offspring [20]. Nevertheless, it is not enough to estimate the possibility that DGATs may be a target for regulating DEHP-induced hepatic lipid accumulation because a single concentration was used.
In addition, excess production of free radicals, such as reactive oxygen species (ROS), may be linked to the development of hepatic steatosis [29]. As free radicals may adversely affect cell survival due to membrane damage through the oxidative damage of lipids and proteins, as well as irreversible DNA modification, the elevation of oxidative stress may cause cellular and molecular damage, thereby causing tissue destruction [30]. Elevation of oxidative stress and inflammation in the liver due to DEHP exposure may influence NAFLD progression [18, 21]. Notably, transcriptome analysis suggested that DEHP disrupts cholesterol by increasing oxidative stress in rat liver [31].
Therefore, this study focused on the association between the levels of DGATs and the disturbance of hepatic lipid metabolism following short-term exposure to DEHP. To understand DEHP-induced hepatic impairment, the mRNA and protein expression of DGAT enzymes and markers of oxidative stress and inflammation were measured in the liver of male adolescent rats.
Materials and Methods
Animals
The experiments in this study and the care of animals complied with the Good Laboratory Practice guidelines for animal experiments defined by the Korea Testing and Research Institute (TBH-1252, 2009/08). Four-week old male SpragueDawley (SD) rats (n=25) bred in the specific pathogen-free status were purchased from Orient Bio (Seongnam-si, Gyeonggido, Korea). To reduce the influence of steroid hormone, only male SD rats were used. The rats were maintained under controlled environmental conditions (temperature: 23 ± 3 °C, relative humidity: 55±15%, and light/dark cycle of 12 h: light from 20:00 to 08:00 h) and provided with free access to appropriate food (Purina Korea Inc., Gyeonggi-do, Korea) and distilled water. After acclimatization to the light/dark cycle for one week, the rats were divided into five groups (control [corn oil], DEHP 0.75, 7.5, 15, or 150 mg/kg/day) with five rats per group.
Dose Selection
Female rats exposed to 7.5 mg/kg body weight DEHP on alternate days for 14 days showed a decrease in serum insulin and increase in blood glucose [32]. In our previous study, the increase of TG levels and lipid droplets in the liver after perinatal exposure to DEHP 0.75 mg/kg/day were observed in adult male offspring [20]. Thus, we selected 0.75 and 7.5 mg/kg body weight/day DEHP to examine the impact of short-term exposure to DEHP on hepatic lipid metabolism. In addition, higher (15 mg/kg body weight/day) concentrations were also used to evaluate the plausible influence of short-term exposure of DEHP on lipogenesis. The highest dosage of DEHP, 150 mg/kg/day, was used as the positive control.
Experimental Design
DEHP (purity 99 %, CAS no. 117-81-7) and corn oil (CAS no. 8001-30-7) were purchased from Sigma Chemical Company (St. Louis, MO, USA). Varying concentrations of DEHP (0.75, 7.5, 15, and 150 mg/kg/day) were prepared by dissolving DEHP in corn oil. Administration volume did not to exceed 2 mL per administration based on the maximum of 10 mL/kg. Rats were administered corn oil or DEHP once daily by oral gavage for one week. At the end of the experiment, all rats were anaesthetized with isophorone after 12 h of fasting. Blood samples were obtained from the vena cava using a needle (21 to 25 G), and then centrifuged for 15 min (1,000 ×g at 4 °C) to separate the serum. Serum was distributed into sterile cryovials (Greiner Labortechnik, Frickenhausen, Germany) in volumes of 500 μL and immediately frozen at –80 °C for performing biochemical, cytokine, and malondialdehyde (MDA) analyses. The dissected livers were rinsed with ice-cold phosphate-buffered saline (PBS). After measuring the weight of the liver, some samples were fixed in formalin for histological observation and frozen in a sterile cryovial (Greiner Labortechnik, Frickenhausen, Germany) for MDA, TG, and molecular analyses.
Body Weight and Relative Liver Weight
The body weight of SD rats was measured on the first and last day (seventh day) of treatment. The liver weight was measured after necropsy and the relative liver weight (%) was calculated as absolute liver weight (g)/body weight (g) × 100.
Hormone and Cytokine Levels in the Serum and Liver
Serum leptin (IBL Japan, Gunma, Japan; 27295), insulin (Mercodia AB, Uppsala, Sweden; 10-1124-01), adiponectin (Adipogen Inc., Incheon, Korea; AG-45A-0005EK-KI01), hepatic interleukin-6 (Thermo Fisher Scientific, Inc., Waltham, MA, USA; BMS625), and tumour necrosis factor-α (Thermo Fisher Scientific, Inc., Waltham, MA, USA; KRC3011) were measured using enzyme-linked immunosorbent assay kits according to the manufacturer’s protocol. The absorbance was measured using a plate reader (Tecan Sunrise TW, Salzburg, Austria).
Malondialdehyde (MDA) levels in the Serum and Liver
The frozen liver samples were homogenized in deionized water and minced samples were sonicated for 10 s and frozen/thawed at ≤ - 20 °C (repeated three times). The serum and homogenized liver samples were aliquoted to a microcentrifuge tube (300 μL) and thiobarbituric acid reactive substances acid reagent (300 μL) was added and mixed well. The samples were incubated for 15 min at room temperature and centrifuged for 4 min (≥ 12000 ×g, at 4 °C). The supernatants were collected to measure MDA levels. The preparation and measurement of MDA in the serum and liver were performed according to the manufacturer’s protocol (R&D Systems Inc., Minneapolis, MN, USA; KGE013). The absorbance (532 nm) was measured using a plate reader (Tecan Sunrise TW, Salzburg, Austria).
TG Contents in the Liver
Frozen liver samples were homogenised in PBS (1:9, w/v) on ice and sonicated to further break the cell membranes. Minced tissues were centrifuged for 5 min (5000 ×g, at 4 °C). The supernatants were collected to measure the hepatic TG levels. The preparation and measurement of TG content in the liver samples were performed according to the manufacturer’s protocol (MyBiosource, Inc., San Diego, CA, USA; MBS164762). Absorbance (450 nm) was measured using a plate reader (Tecan Sunrise TW, Salzburg, Austria).
Histological Observations
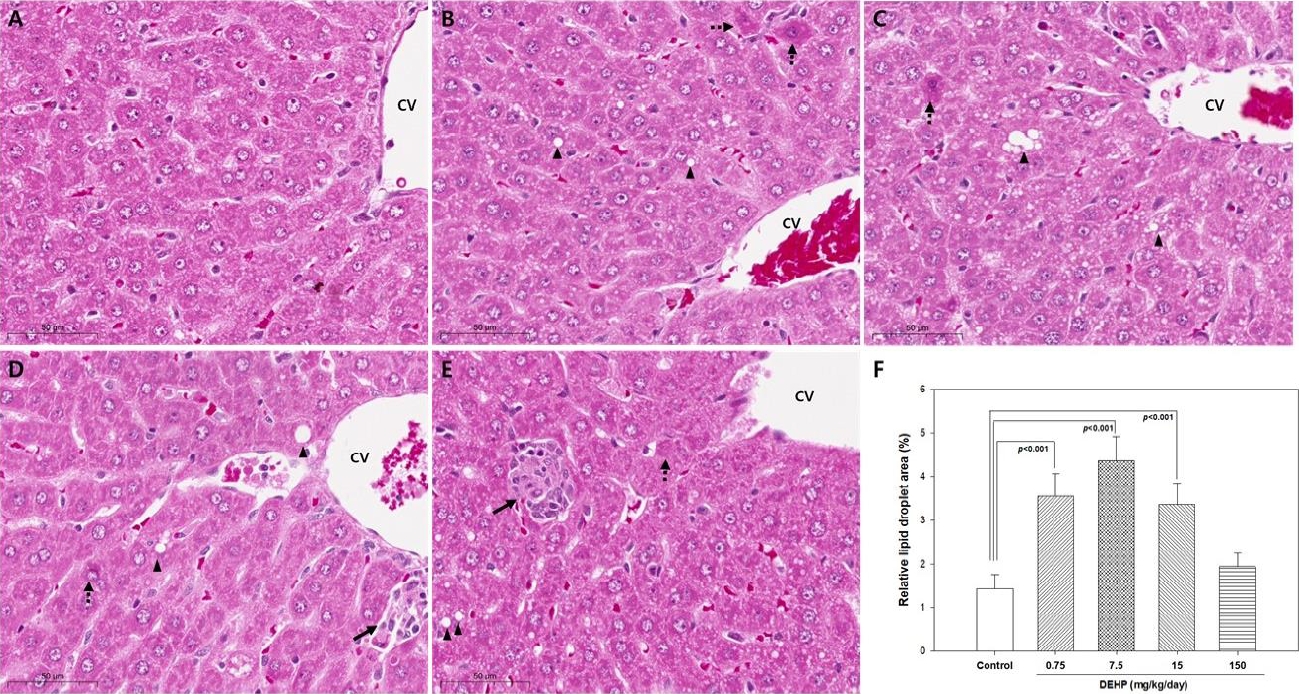
The liver was preserved and fixed in 10% neutral solution buffered with formalin. The fixed liver samples were dehydrated with graded ethanol and embedded in paraffin. The embedded tissues were sectioned (5μm thickness) and stained with periodic acidschiff and hematoxylin-eosin. The stained section were examined for the presence of tissue damage using a light microscope (Olympus Japan Co., Tokyo, Japan). Ten non-overlapping random fields per mouse were acquired using a Fuji Digital Camera HC300Z/CL (Olympus Japan Co., Tokyo, Japan), and the percentage of lipids in the total liver section was assessed using QuPath 0.4.3 software (University of Edinburgh, Edinburgh, Scotland) [33].
Quantitative Real-time PCR (qRT-PCR) Analysis of Liver Samples
Frozen liver samples (≤ 30 μg) were homogenized and isolated to extract the total RNA using an RNA isolation kit (Qiagen, Hilden, Germany; 74004) according to the manufacturer's protocol. First-strand cDNA was synthesized from isolated total RNA using the PrimeScript™ RT Master Mix (Takara, Otsu, Shiga, Japan; RR036A). Analysis of the mRNA expression of target genes was performed using qRT-PCR. Relative expression levels were determined using the ΔΔCt method. qRT-PCR was performed in duplicate using the Thermal Cycler Dice ® real-time system (Takara, Otsu/Shiga, Japan). The average expression levels of rat glyceraldehyde-3-phosphate dehydrogenase (Gapdh), β-Actin, and ribosomal protein lateral stalk subunit P0 (Rplp0) were used to normalize the expression levels of the target genes. Primer sequences used are listed in Table 3. The protocol of RT-PCR reactions included an initial denaturation (95 °C for 2 min, 1 cycle), denaturation (10 sec each at 95 °C, 45 cycles), and annealing and extension (40 sec at 62 °C).
Western Blot Analysis of Liver Samples
Frozen liver samples were homogenized using cold radioimmunoprecipitation assay buffer (Sigma-Aldrich Co. LLC., St. Louis, MO, USA; R0278) and centrifuged for 10 min (760 ×g, 4 °C). The supernatant was collected to measure the total protein concentration using the Bradford protein assay (Thermo Fisher Scientific Inc., Waltham, MA, USA; 23225). Protein aliquots (each 50 μg/well) were separated on an 8–12% sodium dodecyl sulphate-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (GE Healthcare Life Sciences, Pittsburgh, PA, USA). The membranes were incubated with 5% (w/v) skim for about 2 h for blocking. After blocking, the membranes were incubated with primary antibodies against GAPDH (Santa Cruz Biotechnology Inc., Dallas, TX, USA; sc-32233, 1:2000), DGAT1 (Novus Biologicals, Littleton, CO, USA, NB110-41487, 1:1000), and DGAT2 (Novus Biologicals, Littleton, CO, USA, NBP1-71701, 1:1000) diluted in Tris-buffered saline-Tween 20 (TBS-T) at 4 °C overnight. The membranes were washed three times with TBS-T for about 10 min each and then incubated with secondary anti-rabbit IgG (Santa Cruz Biotechnology Inc. Dallas, TX, U.S.A., sc-2004, 1:4000) antibody at room temperature for 40 min. After incubating with the secondary antibody, the membranes were three times washed with TBS-T for about 10 min each. The images were obtained using an LAS 4000 instrument (GE Healthcare Bio-Sciences Corp., Piscataway, NJ, USA). To quantify the density of each band, the ImageQuant TL software (GE Healthcare Bio-Sciences Corp. Piscataway, NJ, USA) was used.
Statistical Analysis
Data (body and relative liver weight, as well as hormone, cytokine, mRNA, and protein levels) are expressed as the mean ± standard error. The Shapiro–Wilk test was performed to test for normality. Given the outliers and the small sample size, the distribution was not normal. Thus, Kruskal–Wallis test was used for comparisons among groups. Dunn’s test was performed to determine which groups were different even if the association was not statistically significant between DEHP exposure and the outcomes. Data analysis was performed using STATA (version 16.0 StataCorp LP College Station, TX, USA). P <0.05 was considered significant.
Results
Body Weight, Relative Liver Weight, and Hormone and Cytokine Levels
The initial average body weights were similar between the groups (Table 2). The average body weight gain (g) and relative liver weight of the DEHP 150 mg/kg/day group were significantly higher than those of the control group (P = 0.044 and P = 0.049, respectively). Serum insulin levels in the DEHP 7.5 mg/kg/day group were significantly higher than those in the control group (P = 0.027). Serum leptin levels in the 15 and 150 mg/kg/day DEHP groups were significantly higher than those in the control group (P = 0.046 and P = 0.022, respectively). There were no statistically significant differences in the adiponectin and TG concentrations between the control and DEHP-treated groups.
The average hepatic TG level in the control group was 2.84 ± 0.13 mg/g protein. Rats treated with DEHP 0.75 mg/kg/day tended to have decreased TG levels in the liver compared to control rats; however, this was not statistically significant (2.38 ± 0.16 mg/g protein). Rats treated with DEHP 7.5, 15, and 150 mg/kg/day did not show significant changes in the hepatic TG levels compared to control rats.
Oxidative Stress Parameter and Inflammatory Cytokines Concentrations
The serum levels of MDA, a marker of oxidative damage, were significantly elevated in the DEHP 7.5 and 150 mg/kg/day groups compared with those in the control group (both P=0.026; Table 3). In addition, 7.5, 15, and 150 mg/kg/day DEHP significantly increased the MDA levels in the liver compared with those in the control group (P=0.017, P=0.001, and P = 0.002, respectively).
Histological Observation in the Liver
In the control group, the central vein was surrounded by normal hepatocytes and hepatic parenchyma (Figure 1A). The DEHP 0.75, 7.5 and 15 mg/kg/day treated groups showed significant increase in lipid droplets in the liver compared to those in the control group (all P<0.001; Figure 1F). Inflammatory infiltrates near the central vein were observed in rats treated with 7.5, 15, and 150 mg/kg/day DEHP (Figure 1C – 1E). In addition, acidophilic bodies were occasionally observed in rats treated with DEHP treated groups (Figure 1B - 1E).
DGAT1/2 mRNA and Protein Expression in the Liver
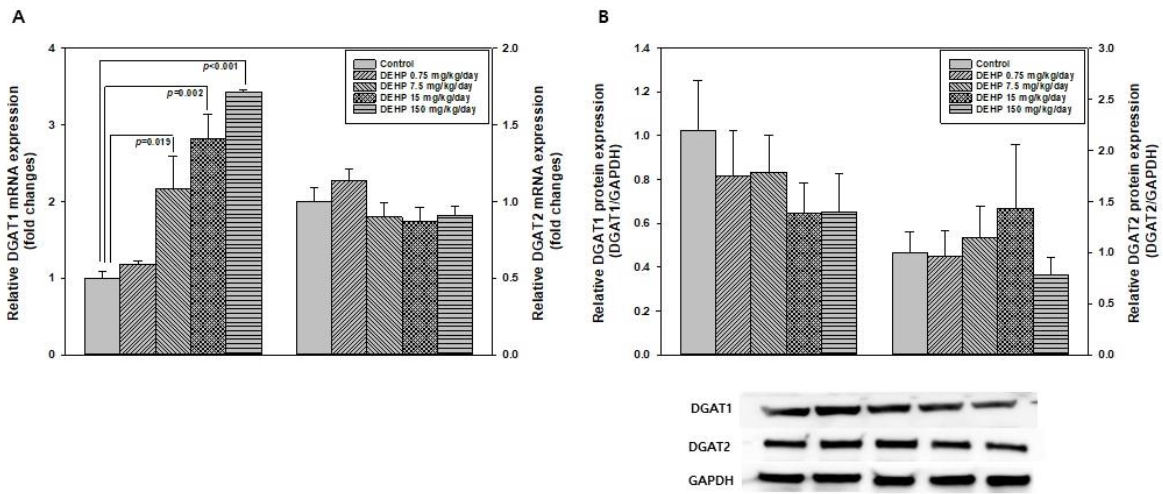
The expression of Dgat1 in the liver was significantly increased in DEHP 7.5, 15, and 150 mg/kg/day groups compared with those in the control group (P = 0.019, P = 0.002, and P < 0.001, respectively [Figure 2A]). The expression of Dgat2 was similar between the control and DEHP-treated groups. However, the protein expression levels of DGAT1 and DGAT2 did not show a significant difference between control and DEHP-treated groups (Figure 2B).
Discussion
This study investigated the role of DGATs in hepatic lipid metabolism following DEHP exposure. The Dgat1 expression in the liver significantly increased as the DEHP concentration increased, but the DGAT1 protein levels did not differ between the control and DEHP-treated groups. MDA levels in the serum and liver were significantly elevated in the DEHP-treated groups compared to that in the control group. In accordance with the MDA levels, portal inflammatory infiltrates and acidophilic bodies were observed in the liver of rats treated with DEHP at 7.5 mg/kg/day and above. Based on the histological quantification, a relative increase in lipid accumulation was observed in DEHP 0.75, 7.5, and 15 mg/kg/day-treated groups compared to that in the control group.
DEHP is commonly used to increase the flexibility of plastic products [1]. It can enter the human body through ingestion, inhalation, and dermal contact in daily life (ranging from 5–100 μg/kg body weight/day) [2]. In patients using medical devices such as hemodialysis and blood transfusion, the exposure levels may increase up to 8 mg/kg body weight/day in adults [2]. This is higher than the no-observed-adverse-effect-level of DEHP (5 mg/kg body weight/day) based on testicular and reproductive toxicity [34]. Despite not reflecting an environmental relevant level, exposure to DEHP at extremely high doses may occur, although rarely. Thus, instead of using a relatively higher dose than the environmental exposure levels, the exposure duration was limited to 7 days in this study.
Several studies have shown that DEHP can potentially damage the reproductive system [35-37]. Recently, a plausible association between DEHP and metabolic disorders, including obesity, diabetes mellitus, and thyroid dysfunction, has been suggested [32, 38, 39]. In addition, the disruption of hepatic lipid metabolism was observed in DEHP treated with sufficient fats [18, 21-23]. Although abundant fat supply might be important in the development of NAFLD, our previous study showed that perinatal exposure to DEHP with a normal diet induced hepatic lipid accumulation in adult male offspring [20]. Similar with our results, DEHP-exposed adult male zebrafish showed the disruption of genes expression related to the metabolic processes in the liver [19]. In this study, relative lipid droplet areas in the liver (%) were significantly increased in DEHP treated groups, except at the highest concentration, compared to those in the control group. This shows that DEHP itself may affect NAFLD progression.
The significant increase in enzymes related to fatty acid synthesis, such as peroxisome proliferator-activated receptors (PPARs) and sterol regulatory element-binding protein 1, following DEHP treatment in rats [18, 21] and HepG2 cells, [21] might represent that DEHP could influence lipid synthesis and liver metabolism. In addition, the activation of PPARγ resulting from DEHP exposure might induce oxidative stress and reduce insulin receptor and glucose transporter 4 expression in normal human hepatocytes [22]. Thus, DEHP may influence lipid metabolism in the liver through elevation of PPARs expression. However, as PPARs upregulation does not occur in humans [40], it might be difficult to extrapolate the action of DEHP from animal models to humans.
DGAT enzymes are involved in the final step of converting diacylglycerol to TG in the liver [24, 41]. DGAT1 is mainly present in the endoplasmic reticulum, whereas DGAT2 is present in the endoplasmic reticulum and around lipid droplets [25]. DGAT2 is considered a dominant enzyme in TG synthesis homeostasis, because approximately 90 % of DGAT2 deficient mice have decreased TG levels in the liver and die shortly after birth [42]. Overexpression of either DGAT1 or DGAT2 increased the TG content in plant, insect, or mammalian cell lines [24, 43], and increased the de novo synthesis and accumulation of TG [42]. However, DGAT enzymes have different DNA and protein sequences and functions, and cannot complement each other’s roles. When DGAT1 is active, relatively small lipid droplets are formed [44, 45] whereas, large lipid droplets (typically 1–2 μm) are formed in DGAT2-expressing cells [42, 45]. Our previous study demonstrated that perinatal exposure to DEHP could influence susceptibility to NAFLD in male offspring through upregulation of DGAT1 in the liver [20]. In this study, compared to the control group, there was a marked increase in Dgat1 mRNA expression in the liver after DEHP treatment. However, there were no differences in the DGAT1 protein expression in the liver between the control and DEHP-treated rats. As DGAT1 is responsible the re-esterification of free fatty acids to diacylglycerol to form TGs during lipolysis, the absence of difference in DGAT1 protein expression between control and DEHP treatment groups might be linked to the lack of TG accumulation in the liver. In addition, the discrepancy result of DGAT expression in the liver might be the increase of oxidative stress caused by relatively high dosages of DEHP.
Oxidative stress can be caused by various environmental chemicals including DEHP. Because the elevation of oxidative stress subsequently enhances the release of lipid peroxidation and causes oxidative modification of biomolecules in cells, such as DNA, lipids, and proteins, oxidative stress is related with liver damages [29, 30, 46]. In vitro and in vivo studies have been shown that exposure to DEHP-induced the excess oxidative stress, such as MDA and the decrease of antioxidant enzymes, such as superoxide dismutase in human liver cancer cell line and in rat liver [18, 21, 31]. Quails treated with DEHP showed lipid metabolism disorder and inflammatory response in the liver resulted from the activation of LXR/SREP-1c, PPARα/γ, and NF-kB signal pathways [47]. In this study, the lowest dose of DEHP showed no significantly increase in oxidative stress in the serum and liver, but hepatic lipid amounts measured by a quantitative method showed a significant increase compared to the control group. However, as the dose of DEHP increased, MDA levels in the serum and liver, an indicator of oxidative stress, increased significantly and histological changes such as inflammation and acidophilic bodies were observed in the liver. In particular, at the highest dose of DEHP, histological changes were observed without lipid accumulation in the liver. It seems that relatively high dosages of DEHP do not promote hepatic lipid accumulation, but rather induce oxidative stress, which promotes inflammation and fibrosis and consequently progresses to liver injuries. Although the molecular mechanism of liver injuries induced by DEHP has not yet been clearly identified, the management of oxidative stress may help to control the progression of DEHP-induced liver damages. Because the nuclear factor E2-related factor 2 (Nrf2) is regulated genes expression related with detoxifying enzymes, redox proteins, and lipid metabolism [48], the modulation of Nrf2 can improve an antioxidant status, and prevent and treatment of liver disease [49]. Recently, DEHP-induced liver toxicities were improved after lycopene, which found in red foods, in mice [50]. It seems that the regulation of Nrf2-mediated pathway using antioxidants may prevent DEHP-induced liver damage. However, due to the use of higher dose than that in the environment, it is difficult to predict the effects in animals treated with DEHP at environmental relevant levels. Our results show the increase of oxidative stress at levels of DEHP that can be exposed by using PVC-containing medical devices, thereby it is needed the evaluation of antioxidant capacity on hepatic lipid metabolism caused by DEHP
Therefore, short-term exposure to DEHP may disrupt hepatic lipid metabolism by increasing the oxidative stress and inflammatory response rather than lipid accumulation. In addition, excessive increase of oxidative stress may interfere with the translational activity of DGAT1, which has an important role in TG storage in the liver. Additional experiments should be performed to examine the specific role of oxidative stress induced by long-term exposure to DEHP at environmental relevant levels on the expression of DGAT enzymes and related pathway of TG synthesis in the liver.
Conclusions
DEHP may affect hepatic lipid metabolism through mediating DGAT1 expression. When DEHP-induced excessive oxidative stress, the expression of DGAT1 might suppressed, and hepatotoxicity could be caused. Therefore, it is necessary to evaluate the influence of oxidative stress induced by DEHP exposure on Dgat1 translation in the liver. The relatively short period of DEHP treatment (one week) and the small number of subjects in each group (n=5/group) could be considered study limitations. Further studies are needed to identify the role of ROS caused by long-term exposure to DEHP.